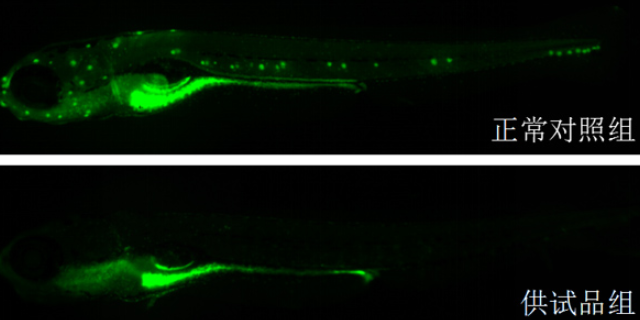
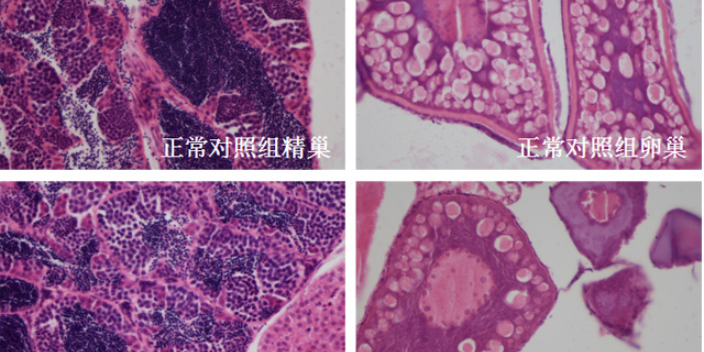
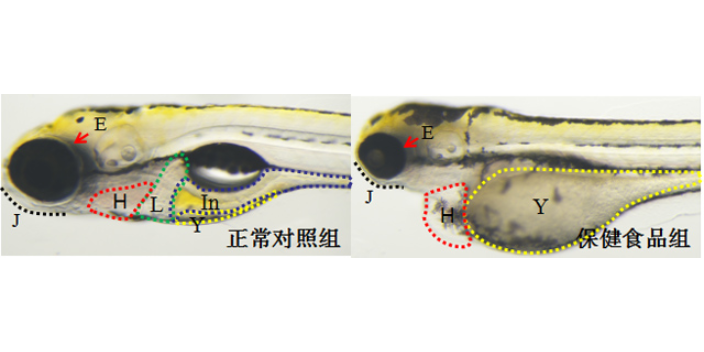
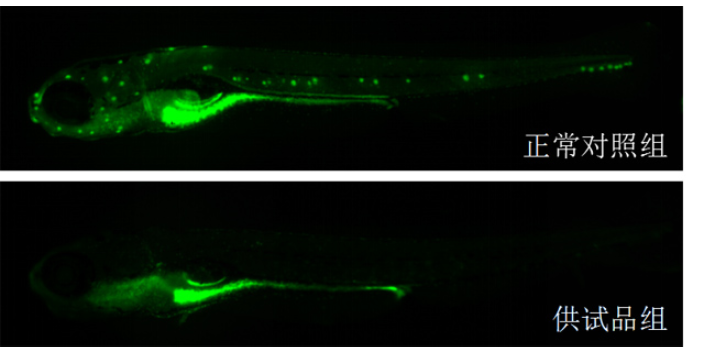
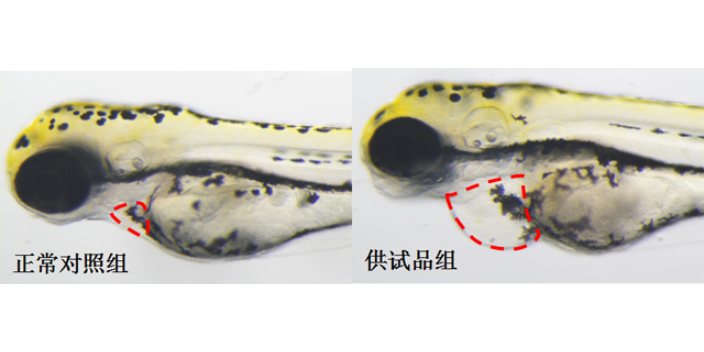
体外模型是生物大分子临床前研究的首要环节,主要用于靶点结合亲和力测定、细胞水平活性验证及作用机制解析。表面等离子共振(SPR)技术可实时监测抗体与抗原的动态结合过程,量化KD值(解离常数),例如PD-1/PD-L1抑制剂的筛选中,SPR能精细区分不同抗体亚型的结合强度。细胞水平实验则通过报告基因系统(如NF-κB荧光素酶报告基因)或流式细胞术,评估抗体对信号通路的影响或抑制效应。例如,在EGFR突变型肺ancer药物开发中,体外3Dtumor球体模型可模拟tumor微环境,验证抗体对细胞增殖、凋亡及血管生成的影响。此外,类organ技术通过患者来源tumor组织培养,为生物大分子提供更贴近临床的个体化评价平台,其预测药物敏感性的准确率较传统2D细胞模型提升30%以上。临床前实验降低研发风险,环特生物定制化设计实验方案。深圳免疫药物临床前

生物大分子药物(如抗体、蛋白、核酸等)因其高特异性和强的效性,已成为现代医药研发的关键方向。然而,其临床前研究面临独特挑战:分子量大导致膜通透性差、免疫原性风险高、稳定性控制难,且需针对特定靶点设计复杂作用机制。例如,单克隆抗体需通过抗体依赖细胞介导的细胞毒性(ADCC)或补体依赖细胞毒性(CDC)发挥作用,而双特异性抗体则需同时结合两个抗原表位以实现精细调控。临床前阶段需系统评估这些分子的药代动力学(PK)、药效动力学(PD)及毒性特征,通常采用体外细胞模型(如HEK293、CHO细胞)和体内动物模型(如小鼠、非人灵长类)相结合的策略。数据显示,全球生物大分子药物临床前研发失败率高达40%,其中因免疫原性或药代动力学问题导致的淘汰占比超60%,凸显了临床前研究的重要***物临床前安全性试验临床前实验需严谨设计,环特生物拥有标准化实验体系.

毒代动力学(TK)研究通过测定动物体内药物浓度-时间曲线,明确毒性剂量下的暴露量(AUC、Cmax),为毒性机制解析提供剂量依据。例如,某肝毒性的药物在重复给药毒性实验中,发现300mg/kg剂量下肝酶升高,TK研究显示该剂量下血药浓度是疗效剂量的10倍,提示毒性源于过度暴露。风险评估则结合毒理学数据与临床预期暴露量,计算安全边际(MarginofSafety,MOS=NOAEL/临床剂量)。若MOS≥10,认为安全性可控;若MOS<5,则需重新优化结构或调整给药的方案。此外,基于生理的药代动力学模型(PBPK)可预测不同人群(如儿童、肝肾功能不全患者)的毒性风险,为个性化用药提供依据。终,毒理学研究需形成综合报告,明确“可接受风险”与“需关注风险”,支持IND申报及临床试验设计。
随着医药创新与监管趋严,临床前研究对模型真实性、数据规范性、评价全面性提出更高要求,环特生物持续升级临床前技术能力,打造更贴合临床转化需求的临床前研究体系。公司在临床前研究中引入类organ、PDX、基因编辑等前沿技术,构建高度模拟人体病理生理状态的临床前评价模型,提升实验结果向临床转化的预测准确率。在临床前药效评价方面,环特针对tumor、神经退行性疾病、代谢疾病、炎症、心血管疾病等多个医疗领域,建立标准化临床前药效评价方案,可完成tumor抑制、神经保护、降糖降脂、抑炎、血管生成等多维度药效检测。在临床前安全性评价方面,公司配备专业病理分析、影像追踪、生物标志物检测平台,实现对靶organ毒性、免疫反应、代谢特征的精细解析。环特生物坚持以临床前研究数据为关键,以法规合规为准绳,帮助企业在研发早期识别风险、优化结构、提升成药的性,为后续临床试验设计提供科学依据,推动质量候选药物更快进入临床阶段。临床前研究可有效降低新药研发风险,提升研发成功率。

候选成药分子的临床前研究是药物开发链条中的关键环节,其关键目标是通过系统评估分子的安全性、有效性及药代动力学特性,为后续临床试验提供科学依据。这一阶段需回答三个关键问题:分子是否具备医疗潜力?是否安全可控?能否在目标组织中达到有效浓度?研究内容涵盖体外活性筛选(如酶抑制、细胞增殖实验)、体内药效验证(如疾病动物模型)、毒理学评估(急性/慢性毒性、遗传毒性)及药代动力学(ADME:吸收、分布、代谢、排泄)分析。例如,针对阿尔茨海默病的候选分子Aβ寡聚体抑制剂,临床前需在转基因小鼠模型中验证其能否改善认知功能,同时通过肝微粒体孵育实验评估其代谢稳定性。这一阶段的成功标准是获得“安全有效”的初步证据,支持向IND(新药临床研究申请)申报迈进,其决策准确性直接影响药物开发成功率(据统计,临床前研究充分的分子进入临床后的成功率可提升40%)。杭州环特生物专注优化临床前医药研究的技术流程。上海生物药临床前模式动物
临床前医药研究是连接药物研发与临床应用的桥梁。深圳免疫药物临床前
生物大分子临床前研究的后续目标是实现从实验室到临床的转化。转化医学通过整合临床前数据与早期临床试验结果,优化药物设计。例如,基于临床前药代动力学模型预测人体剂量,可减少I期临床试验的剂量探索范围。监管科学则聚焦于建立符合国际标准的评价体系,FDA的“动物法则”(Animal Rule)允许在特定情况下(如生物影响袭击药物开发)以动物数据替代临床数据,而EMA的“适应性许可”路径则支持基于早期临床前数据的条件性上市。此外,人工智能(AI)技术正重塑临床前研究范式,通过机器学习算法分析海量临床前数据,可预测药物在人体中的疗效及安全性,例如DeepMind的AlphaFold已用于预测抗体-抗原复合物结构,加速候选分子筛选。未来,随着类器官芯片、单细胞测序等技术的融合,生物大分子临床前研究将迈向更精细、高效的阶段。深圳免疫药物临床前