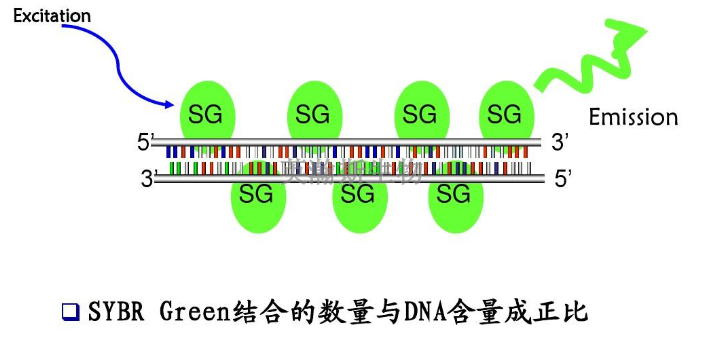
PCR实验技术中cDNA第二链的合成方法有以下几种:1、自身引导法合成的单链cDNA3'端能够形成一短的发夹结构,这就为第二链的合成提供了现成的引物,当***链合成反应产物的DNA:RNA杂交链变性后利用大肠杆菌DNA聚合酶ⅠKlenow片段或反转录酶合成cDNA第二链,***用对单链特异性的S1核酸酶消化该环,即可进一步克隆。但自身引导合成法较难控制反应,而且用S1核酸酶切割发夹结构时无一例外地将导致对应于mRNA5'端序列出现缺失和重排,因而该方法目前很少使用。2、置换合成法该方法利用链在反转录酶作用下产生的cDNA:mRNA杂交链不用碱变性,而是在dNTP存在下,利用RNA酶H在杂交链的mRNA链上造成切口和缺口。从而产生一系列RNA引物,使之成为合成第二链的引物,在大肠杆菌DNA聚合酶Ⅰ的作用下合成第二链。该反应有3个主要优点:(1)非常有效;(2)直接利用链反应产物,无须进一步处理和纯化;(3)不必使用S1核酸酶来切割双链cDNA中的单链发夹环。荧光定量pcr的原理和步骤是什么?新疆组织pcr怎么选择
PCR引物设计的原则PCR反应中有两条引物,即5′端引物和3′引物。设计引物时以一条DNA单链为基准(常以信息链为基准),5′端引物与位于待扩增片段5′端上的一小段DNA序列相同;3′端引物与位于待扩增片段3′端的一小段DNA序列互补。(1)引物设计的基本原则引物长度:15-30bp,常用为20bp左右。引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C过多易出现非特异条带。ATGC比较好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列参照。引物内部不应出现互补序列。两个引物之间不应存在互补序列,尤其是避免3′端的互补重叠。引物与非特异扩增区的序列的同源性不要超过70%,引物3′末端连续8个碱基在待扩增区以外不能有完全互补序列,否则易导致非特异性扩增。引物3‘端的碱基,特别是较末及倒数第二个碱基,应严格要求配对,比较好选择是G和C。引物的5′端可以修饰。如附加限制酶位点,引入突变位点,用生物素、荧光物质、地高辛标记,加入其它短序列,包括起始密码子、终止密码子等。海南推荐pcr实验英瀚斯生物分子生物学平台,配备专业定量pcr仪。

PCR实验技术中的反向PCR(InversePCR)介绍:反向PCR起初设计用于确定临近未知区域的序列。反向PCR有助于研究一个基因的启动子序列;致*染色体重排如基因融合、异位或转移;及病毒基因的整合。之所以称为反向PCR,是因为引物设计用于向两边延伸而不像常规PCR中朝着彼此延伸。如今,反向PCR常用于定点突变,复制一个具有预期突变的目的质粒。在研究基因组DNA未知序列的传统实验流程中,限制性内切酶消化和连接先于反向PCR,接着对PCR扩增子进行测序。对于gDNA消化,需选择一种限制性内切酶进行酶切以获得合适长度可自连的片段。同时,所选择的内切酶不应该切割已知序列,所以连接发生于侧翼的未知序列之间。使用低浓度的酶切DNA的片段以助于片段自连而非多个片段连接。片段自连完成后,通过反向PCR扩增DNA的位置区域。获得的扩增子两端包含一部分已知序列。之后通过测序以鉴定临近已知序列的未知区域。
PCR出现假阳性的原因分析。引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。靶序列太短或引物太短,容易出现假阳性。需重新设计引物。靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。这种假阳性可用以下方法解决:①操作时应小心轻柔,防止将靶序列吸入加样器内或溅出离心管外。②除了酶及其他不耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管均应一次性使用。③必要时,在加标本前,反应管和试剂用紫外线照射,以破坏存在的核酸。二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。荧光定量pcr正常值多少?
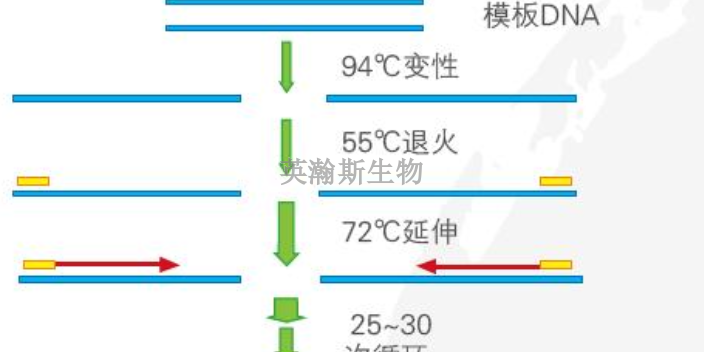
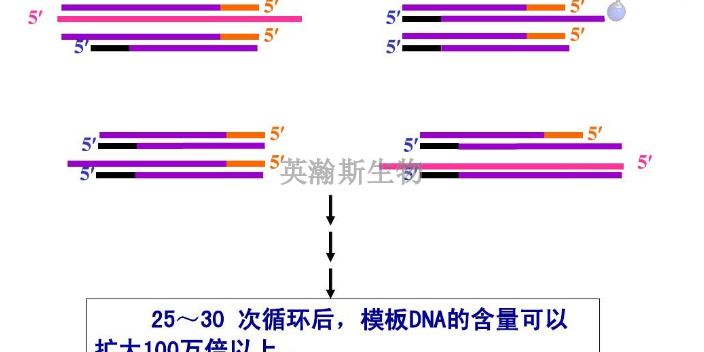
PCR原理:PCR是在试管中进行的DNA复制反应,基本原理是依据细胞内DNA半保留复制的机理,以及体外DNA分子于不同温度下双链和单链可以互相转变的性质,人为地控制体外合成系统的温度,以促使双链DNA变成单链,单链DNA与人工合成的引物退火,然后耐热DNA聚合酶以dNTP为原料使引物沿着单链模板延伸为双链DNA。PCR全过程每一步的转换是通过温度的改变来控制的。需要重复进行DNA模板解链、引物与模板DNA结合、DNA聚合酶催化新生DNA的合成,即高温变性、低温退火、中温延伸3个步骤构成PCR反应的一个循环,此循环的反复进行,就可使目的DNA得以迅速扩增。DNA模板变性:模板双链DNA?单链DNA,94℃。退火:引物+单链DNA?杂交链,引物的Tm值。引物的延伸:温度至70℃左右,TaqDNA聚合酶以4种dNTP为原料,以目的DNA为模板,催化以引物3’末端为起点的5’→3’DNA链延伸反应,形成新生DNA链。新合成的引物延伸链经过变性后又可作为下一轮循环反应的模板PCR,就是如此反复循环,使目的DNA得到高效快速扩增。荧光定量pcr的ct值怎么分析?江苏样本pcr哪家好
细胞上清提取RNA进行PCR检测。新疆组织pcr怎么选择
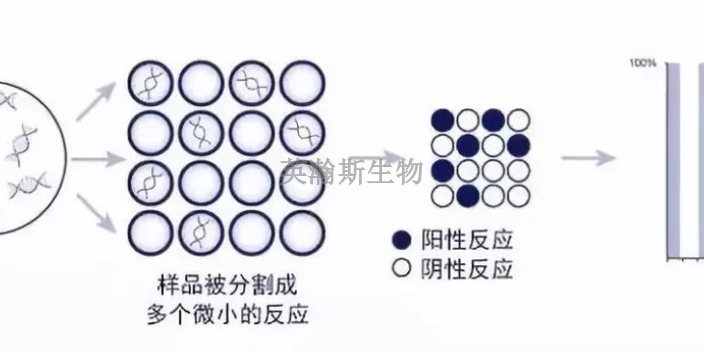
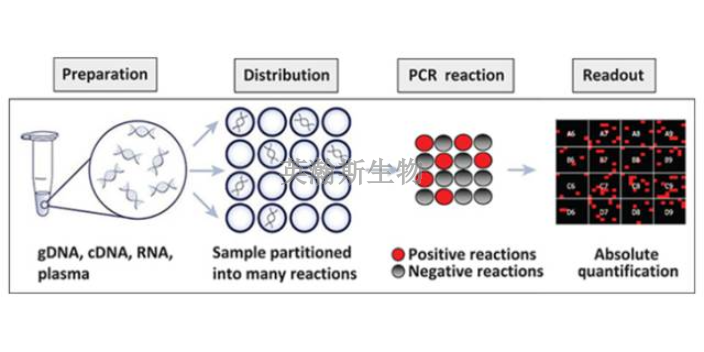
PCR方法主要可以分为三类:终点PCR、qPCR和dPCR。终点PCR终点PCR是较原始、较简单的PCR方法,如今仍在被我们普遍使用。使用者只能在PCR反应结束之后,通过凝胶电泳、毛细管电泳等方法对产物进行检测。终点PCR本身是无法定量的,因为该反应产出的DNA量不一定能反映较初情况。举例来说,不同样品和序列的扩增效率是有差异的。qPCR和dPCR研究者们通过两种途径克服了PCR定量分析的挑战:实时定量PCR(qPCR)和数字PCR(dPCR)。qPCR主要是利用插入性染料或荧光探针(比如TaqMan),人们可以通过监控PCR过程中的荧光强度比较多个样品的DNA水平。数字PCR(dPCR)的基本工作原理很简单,先将样品划分为多个PCR反应,每个反应较多只含有一个模板。然后通过计数正负反应来确定初始样品中模板分子的数量。新疆组织pcr怎么选择
南京英瀚斯生物科技有限公司是一家医学实验外包、动物实验外包、细胞实验外包、分子实验检测、病理染色检测、科研实验外包、动物模型构建、第三方检测、病理组织切片、细胞培养、电生理检测、医学研究和试验发展、生物医药技术研发、技术转让、技术咨询及技术服务的公司,致力于发展为创新务实、诚实可信的企业。英瀚斯生物作为医学实验外包、动物实验外包、细胞实验外包、分子实验检测、病理染色检测、科研实验外包、动物模型构建、第三方检测、病理组织切片、细胞培养、电生理检测、医学研究和试验发展、生物医药技术研发、技术转让、技术咨询及技术服务的企业之一,为客户提供良好的实验外包,动物模型构建,细胞分子实验,病理检测。英瀚斯生物始终以本分踏实的精神和必胜的信念,影响并带动团队取得成功。英瀚斯生物创始人俞东源,始终关注客户,创新科技,竭诚为客户提供良好的服务。