生物大分子药物(如抗体、蛋白、核酸等)因其高特异性和强的效性,已成为现代医药研发的关键方向。然而,其临床前研究面临独特挑战:分子量大导致膜通透性差、免疫原性风险高、稳定性控制难,且需针对特定靶点设计复杂作用机制。例如,单克隆抗体需通过抗体依赖细胞介导的细胞毒性(ADCC)或补体依赖细胞毒性(CDC)发挥作用,而双特异性抗体则需同时结合两个抗原表位以实现精细调控。临床前阶段需系统评估这些分子的药代动力学(PK)、药效动力学(PD)及毒性特征,通常采用体外细胞模型(如HEK293、CHO细胞)和体内动物模型(如小鼠、非人灵长类)相结合的策略。数据显示,全球生物大分子药物临床前研发失败率高达40%,其中因免疫原性或药代动力学问题导致的淘汰占比超60%,凸显了临床前研究的重要性。临床前实验覆盖多维度检测,环特生物实现一站式技术支持.nmpa临床前药物剂量探究
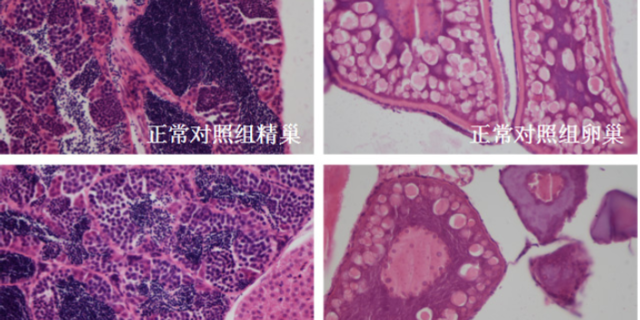
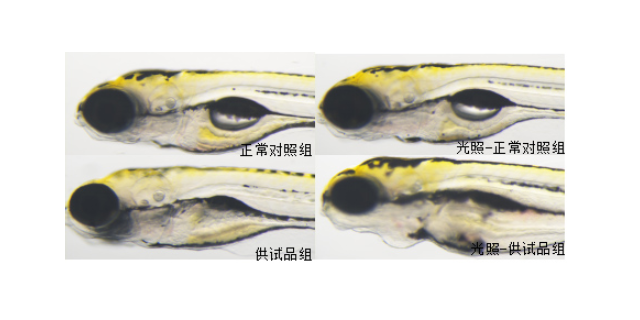
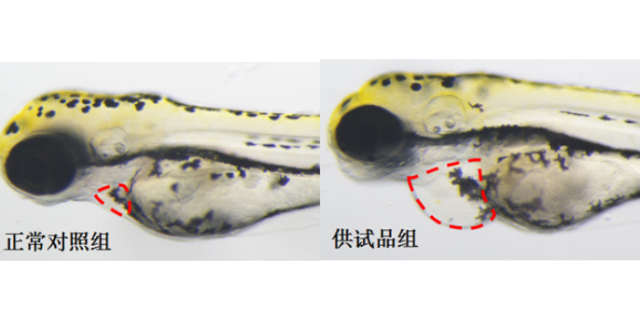
毒理学评价旨在识别药物的潜在毒性,包括急性毒性、慢性毒性、遗传毒性及生殖毒性等。传统方法依赖大鼠、犬等哺乳动物模型,但存在周期长、成本高的局限。斑马鱼模型因其胚胎透明、发育快速,成为急性毒性筛选的优先。例如,OECD指南将斑马鱼胚胎急性毒性测试(FET)纳入标准方法,通过观察胚胎死亡率、畸形率,72小时内可完成初步评估。遗传毒性评价采用Ames试验(细菌回复突变)或小鼠淋巴瘤细胞试验,检测药物是否诱发基因突变。生殖毒性研究则通过斑马鱼胚胎发育毒性测试,评估药物对心脏、神经系统的发育影响。环特生物建立的“斑马鱼-类organ”联合毒理平台,可模拟药物对肝、肾等organ的特异性损伤,提高毒性预测的准确性。例如,某候选药物在类organ中显示肝细胞凋亡率升高30%,而在斑马鱼模型中观察到肝区荧光减弱,两者结合提示需调整剂量或结构。北京成都中药临床前评价机构环特生物持续创新,不断提升临床前实验的效率与准确度。

毒理学研究是临床前研究的“安全阀”,需通过急性毒性(单次高剂量给药)、重复给药毒性(28天/90天多次给药)、遗传毒性(Ames试验、小鼠淋巴瘤试验)及生殖毒性(胚胎致死性、致畸性)实验,多方面评估分子的安全性。例如,针对抗凝血药物,需通过大鼠尾静脉出血时间实验确定耐受剂量(MTD),避免引发过度出血风险;针对抗tumor药物,则需关注骨髓抑制(通过血常规检测白细胞、血小板计数)及肝毒性(通过ALT/AST酶活性测定)。特殊毒性研究(如光毒性、心脏毒性)也不可忽视——例如,通过hERG通道抑制实验评估药物是否可能引发QT间期延长。毒理学数据的解读需结合医疗窗(有效剂量与毒性剂量的比值),若候选分子的医疗指数(TI)>5,则认为安全性可控;若TI<3,则需重新优化结构或调整给药的方案。
对新药临床前毒理学研究结果的准确分析与解读直接关系到新药研发的决策。在分析毒理学数据时,首先要关注各项观察指标的变化趋势,不仅只是某个时间点的数值。例如在长期毒性试验中,体重变化曲线若呈现持续下降趋势,可能意味着药物对动物的营养代谢产生了不良影响,需要进一步深入分析原因。同时,要对不同剂量组之间的数据进行对比,判断剂量 - 反应关系是否存在。若随着药物剂量的增加,毒性反应的发生率和严重程度也相应增加,那么这种剂量 - 反应关系的存在提示药物毒性与剂量密切相关。此外,对于一些异常的检测结果,不能孤立地看待,需要综合考虑动物的整体状况、其他相关指标的变化以及试验过程中的各种因素。例如,血液生化指标中某个酶活性的升高,可能是药物直接作用于肝脏细胞导致,也可能是由于动物在试验过程中受到应激等其他因素干扰。只有通过多方面、系统、科学的分析与解读,才能从毒理学研究结果中提炼出真实、准确的信息,为新药能否进入临床试验以及后续的研发方向提供合理的判断依据。临床前医药研究是连接药物研发与临床应用的桥梁。

生物大分子临床前研究的后续目标是实现从实验室到临床的转化。转化医学通过整合临床前数据与早期临床试验结果,优化药物设计。例如,基于临床前药代动力学模型预测人体剂量,可减少I期临床试验的剂量探索范围。监管科学则聚焦于建立符合国际标准的评价体系,FDA的“动物法则”(Animal Rule)允许在特定情况下(如生物影响袭击药物开发)以动物数据替代临床数据,而EMA的“适应性许可”路径则支持基于早期临床前数据的条件性上市。此外,人工智能(AI)技术正重塑临床前研究范式,通过机器学习算法分析海量临床前数据,可预测药物在人体中的疗效及安全性,例如DeepMind的AlphaFold已用于预测抗体-抗原复合物结构,加速候选分子筛选。未来,随着类器官芯片、单细胞测序等技术的融合,生物大分子临床前研究将迈向更精细、高效的阶段。临床前实验结果,为药物剂型优化提供科学指导方向。临床前研究急性毒
环特生物开展基因编辑研究,赋能临床前实验创新升级、。nmpa临床前药物剂量探究
候选成药分子的临床前研究是药物开发链条中的关键环节,其关键目标是通过系统评估分子的安全性、有效性及药代动力学特性,为后续临床试验提供科学依据。这一阶段需回答三个关键问题:分子是否具备医疗潜力?是否安全可控?能否在目标组织中达到有效浓度?研究内容涵盖体外活性筛选(如酶抑制、细胞增殖实验)、体内药效验证(如疾病动物模型)、毒理学评估(急性/慢性毒性、遗传毒性)及药代动力学(ADME:吸收、分布、代谢、排泄)分析。例如,针对阿尔茨海默病的候选分子Aβ寡聚体抑制剂,临床前需在转基因小鼠模型中验证其能否改善认知功能,同时通过肝微粒体孵育实验评估其代谢稳定性。这一阶段的成功标准是获得“安全有效”的初步证据,支持向IND(新药临床研究申请)申报迈进,其决策准确性直接影响药物开发成功率(据统计,临床前研究充分的分子进入临床后的成功率可提升40%)。nmpa临床前药物剂量探究