临床前研究的精细性依赖于实验模型的可靠性,类organ技术与传统模型的协同应用,为临床前研究提供了更贴近人体的实验体系。杭州环特生物科技股份有限公司将类organ技术融入临床前研究服务,与斑马鱼、哺乳动物模型形成互补。类organ作为“微型organ”,能精细模拟人体organ的结构与功能,在临床前药物代谢、毒性评估等方面展现出独特优势,尤其适用于tumor、消化系统疾病等领域的临床前研究。例如在抗tumor药物临床前研究中,类organ模型可重现tumor的异质性,更精细地评估药物对肿瘤细胞的抑制效果;结合斑马鱼模型的快速筛选优势,能实现“筛选-验证”的高效闭环。这种多模型协同的临床前研究模式,大幅提升了实验数据的可靠性与转化价值,为药物研发提供更有力的支撑。环特生物通过 CMA 认证,临床前实验数据具备影响力。北京眼科药临床前安全性评价服务

急性毒性研究通过单次高剂量给药(如口服、静脉注射),测定药物的半数致死量(LD50)或比较大耐受剂量(MTD),明确其急性毒性阈值。例如,某中枢的神经系统药物在大鼠急性毒性实验中,LD50为500mg/kg,而MTD为200mg/kg,提示临床试验起始剂量应低于100mg/kg(通常为MTD的1/2-1/3)。重复给药毒性研究则通过多剂量、长期(如28天、90天)给药,观察靶organ毒性(如肝、肾、心脏)及剂量-毒性关系。以抗纤维化药物为例,在90天重复给药毒性实验中,犬在300mg/kg/天剂量下出现肾小管坏死,而100mg/kg/天剂量下无明显异常,提示临床安全剂量应≤100mg/kg。此类研究需结合病理学(HE染色、免疫组化)和临床病理学(血常规、生化指标)分析,明确毒性靶organ及可逆性(如停药后是否恢复)。云南cro临床前评价完善的质量体系,保障临床前实验全过程符合行业规范。

抑衰老产品市场的快速发展,对产品功效的科学验证提出了更高要求,临床前研究成为抑衰老产品研发的关键环节。杭州环特生物科技股份有限公司构建了多维的抑衰老产品临床前研究体系,从分子、细胞、组织、个体四个层面验证产品的抑衰老功效。在临床前研究中,通过斑马鱼模型评估产品对衰老相关基因表达的影响、对细胞衰老的延缓作用;利用哺乳动物模型检测产品对寿命、运动能力等指标的改善效果。此外,临床前研究还需验证产品的安全性,确保产品在长期使用过程中无潜在风险。环特生物的临床前研究服务,帮助抑衰老产品企业以科学数据支撑产品功效宣称,提升产品市场竞争力,推动行业向规范化、科学化方向发展。
候选成药分子的临床前研究是药物开发链条中的关键环节,其关键目标是通过系统评估分子的安全性、有效性及药代动力学特性,为后续临床试验提供科学依据。这一阶段需回答三个关键问题:分子是否具备医疗潜力?是否安全可控?能否在目标组织中达到有效浓度?研究内容涵盖体外活性筛选(如酶抑制、细胞增殖实验)、体内药效验证(如疾病动物模型)、毒理学评估(急性/慢性毒性、遗传毒性)及药代动力学(ADME:吸收、分布、代谢、排泄)分析。例如,针对阿尔茨海默病的候选分子Aβ寡聚体抑制剂,临床前需在转基因小鼠模型中验证其能否改善认知功能,同时通过肝微粒体孵育实验评估其代谢稳定性。这一阶段的成功标准是获得“安全有效”的初步证据,支持向IND(新药临床研究申请)申报迈进,其决策准确性直接影响药物开发成功率(据统计,临床前研究充分的分子进入临床后的成功率可提升40%)。临床前研究可有效降低新药研发风险,提升研发成功率。
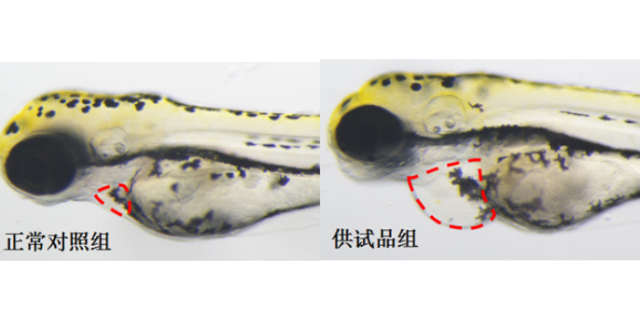
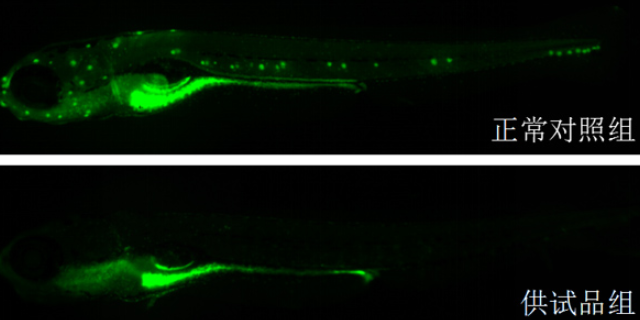
环特生物在靶点发现阶段采用多组学联合分析策略,整合基因编辑、转录组测序及蛋白质组学技术,系统挖掘疾病相关关键靶点。例如,在神经退行性疾病研究中,通过CRISPR/Cas9技术构建斑马鱼α-突触he蛋白过表达模型,结合全脑成像技术,发现特定微小RNA(miRNA)可通过调控自噬通路减缓蛋白聚集,从而锁定miR-34a作为潜在干预靶点。在验证环节,环特利用类organ模型模拟疾病病理特征,例如构建阿尔茨海默病类organ,通过单细胞测序技术揭示Aβ斑块沉积对神经元亚群的影响,为靶点功能验证提供三维组织层面的证据。此外,其开发的斑马鱼荧光报告系统可实时监测靶点活性变化,如Tg(NF-κB:EGFP)转基因斑马鱼通过绿色荧光强度量化炎症信号通路启动程度,加速了靶点验证进程。杭州环特生物专注优化临床前医药研究的技术流程。云南免疫药物临床前安全性评估
环特生物深耕临床前实验,为药物研发筑牢早期研究基础.北京眼科药临床前安全性评价服务
新药临床前毒理学研究是药物开发中保障患者安全的关键环节,其目标是通过系统评估候选药物对实验动物的毒性效应,预测其可能对人体产生的危害,为临床试验的剂量选择、风险控制及后续开发决策提供科学依据。这一阶段的研究需覆盖急性毒性(单次高剂量暴露)、重复给药毒性(多剂量、长期暴露)、遗传毒性(致突变性)、生殖毒性(致畸性、胚胎毒性)及特殊毒性(如光毒性、心脏毒性)等多个维度。据统计,全球约40%的新药在临床前毒理学阶段因安全性问题被淘汰,凸显其“安全阀”作用。例如,某抗tumor候选药物因在犬重复给药毒性实验中发现严重肝坏死,被迫终止开发,避免了潜在的临床肝衰竭风险。毒理学数据的可靠性直接决定了药物能否进入临床试验,其研究设计需严格遵循GLP(良好实验室规范)标准,确保数据的可重复性和监管认可。北京眼科药临床前安全性评价服务