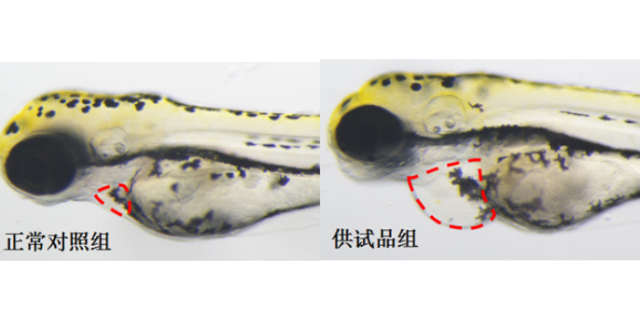
药代动力学(PK)研究聚焦药物在体内的吸收、分布、代谢和排泄(ADME)过程,是决定药物剂量的关键。体外实验中,Caco-2细胞模型可预测药物肠道渗透性,肝微粒体或肝细胞孵育系统则用于评估代谢稳定性。例如,某候选抗ancer药物在肝微粒体中半衰期15分钟,提示需结构优化以提高代谢稳定性。活的体PK研究依赖大鼠或犬模型,通过液相色谱-质谱联用技术(LC-MS)测定血浆、组织中的药物浓度。环特生物开发的斑马鱼PK模型,可实时观察药物在胚胎体内的分布,发现某化合物在脑部的蓄积量是血浆的3倍,提示其可能穿透血脑屏障。PK/PD(药效动力学)整合分析进一步关联药物浓度与疗效,例如在antibiotic研发中,通过PK模型确定给药间隔,使血药浓度维持在小抑菌浓度(MIC)以上,显著提高杀菌效果。杭州环特生物深耕临床前实验领域,为医药研发提供专业技术支撑。浙江国家认可临床前评价

生物制品的临床前安全性评价是药物研发的关键环节,其主要目标在于通过系统化的实验设计,预测药物在人体中的潜在风险,为临床试验提供科学依据。以疫苗为例,其安全性评价需贯穿原辅材料控制、生产工艺验证、理化性质检定、动物试验及临床前监测全流程。动物试验作为主要手段,需模拟人体免疫应答,重点考察疫苗对免疫organ(如胸腺、脾脏)及靶organ(如肝脏、肾脏)的影响,评估毒性可逆性及超敏反应风险。例如,流感疫苗的临床前研究需通过豚鼠主动过敏试验,预测其引发Ⅰ型超敏反应的可能性;而PD-1抑制剂等tumor免疫医疗药物,则需通过非人灵长类动物模型,验证其阻断免疫检查点后的自身免疫风险。评价体系构建需遵循“具体问题具体分析”原则,结合药物作用机制、种属特异性及临床适应症设计试验。对于细胞因子类药物,需考虑其多向性、网络性效应可能引发的“瀑布效应”,如重组人促红的细胞生成素可能同时纠正贫血与促进tumor生长的双重风险。此外,杂质控制是安全性评价的重要环节,宿主细胞蛋白质、DNA残留及内jisu等工艺相关杂质,可能通过免疫复合物沉积导致损伤,需通过纯化工艺优化及质控标准制定降低风险。湖北创新药临床前评价完善的质量体系,保障临床前实验全过程符合行业规范。

动物模型是生物大分子临床前安全性评价的关键环节,需根据药物作用机制选择适宜物种。小鼠模型因其遗传背景清晰、操作便捷,常用于初步药效验证,例如在IL-6抑制剂开发中,通过构建胶原诱导性关节炎(CIA)小鼠模型,可观察抗体对关节肿胀、炎症因子分泌的抑制作用。然而,啮齿类动物与人类在免疫系统、代谢途径等方面存在差异,需进一步通过非人灵长类(NHP)模型进行转化验证。例如,在CD20单抗研发中,食蟹猴模型可更准确预测药物在人体中的半衰期、免疫原性及组织分布特征。此外,转基因动物模型(如人源化FcRn小鼠)通过引入人类基因片段,可模拟生物大分子在人体中的代谢过程,显著提高临床前数据的预测价值。
临床前研究的起点是体外活性筛选,通过高通量技术(如96孔板、自动化液体处理系统)从化合物库中筛选出对靶点具有抑制或活动作用的“苗头化合物”。例如,针对EGFR突变型肺ancer,通过酶联免疫吸附试验(ELISA)筛选能抑制EGFR激酶活性的小分子,初始命中率可能低至0.1%。随后,通过构效关系(SAR)研究优化分子结构——通过合成系列类似物(如改变苯环取代基、调整酰胺键位置),结合表面等离子共振(SPR)技术测定结合亲和力(KD值),逐步提升活性(如将IC50从μM级优化至nM级)。这一阶段需平衡活性与理化性质(如logP、溶解度),避免“活性陷阱”(如过度追求高亲和力导致代谢不稳定)。例如,某候选HER2抑制剂通过引入氟原子降低脂溶性,成功将半衰期从2小时延长至8小时,为后续体内研究奠定基础。临床前实验助力保健食品研发,环特生物提供功效验证。

医疗器械的安全性直接关系到患者生命健康,临床前安全性评价是医疗器械上市前的重要环节。杭州环特生物科技股份有限公司针对医疗器械的特点,提供符合法规要求的临床前安全性评价服务。根据医疗器械的使用场景与接触方式,临床前研究需开展相应的安全性检测,例如植入式医疗器械需进行生物相容性评价、长期毒性测试;体外诊断试剂需进行特异性、灵敏度验证。在临床前研究中,通过斑马鱼模型、哺乳动物模型等,模拟医疗器械在体内的作用过程,评估其对组织organ 的影响;同时,严格遵循ISO、GB等相关标准,确保临床前研究数据的合规性与可靠性。环特生物的临床前安全性评价服务,帮助医疗器械企业满足上市要求,保障产品的临床使用安全。临床前药效学研究可准确评估药物的医疗潜力。浙江化学药临床前实验室
杭州环特生物深耕临床前医药研究,助力新药研发进程。浙江国家认可临床前评价
新药临床前毒理学研究是药物开发中保障患者安全的关键环节,其目标是通过系统评估候选药物对实验动物的毒性效应,预测其可能对人体产生的危害,为临床试验的剂量选择、风险控制及后续开发决策提供科学依据。这一阶段的研究需覆盖急性毒性(单次高剂量暴露)、重复给药毒性(多剂量、长期暴露)、遗传毒性(致突变性)、生殖毒性(致畸性、胚胎毒性)及特殊毒性(如光毒性、心脏毒性)等多个维度。据统计,全球约40%的新药在临床前毒理学阶段因安全性问题被淘汰,凸显其“安全阀”作用。例如,某抗tumor候选药物因在犬重复给药毒性实验中发现严重肝坏死,被迫终止开发,避免了潜在的临床肝衰竭风险。毒理学数据的可靠性直接决定了药物能否进入临床试验,其研究设计需严格遵循GLP(良好实验室规范)标准,确保数据的可重复性和监管认可。浙江国家认可临床前评价