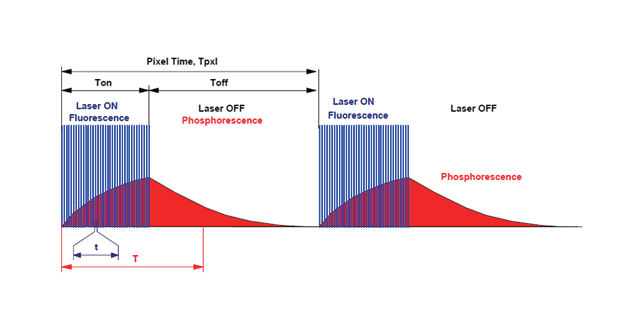
在高光子密度的情况下,荧光分子可以同时吸收两个长波长的光子,然后发射出一个波长较短的光子,其效果和使用一个波长为长波长一半的光子去激发荧光分子是相同的(如下图)。如烟酰胺腺嘌呤二核苷酸(NADH),在单光子激发时,在波长为350nm光的激发下发出450nm荧光;而在双光子激发时,可采用750nm的激发光得到450nm荧光。由于双光子激发需要很高的光子密度,为了不损伤细胞,双光子显微镜使用高能量锁模脉冲激光器。这种激光器发出的激光具有很高的峰值能量和很低的平均能量,从而可以减少光漂白和光毒性带来的不利影响。如果已经有了飞秒光,就可以几套双光子显微镜共享一台,只需分光即可。美国bruker双光子显微镜厂家

n掺杂可以明显影响碳点(CDs)的发射和激发特性,使双光子碳点(TP-CDs)具有本征双光子激发特性和605nm红光发射特性。在638nm激光的照射下,除了长波激发和发射外,还能产生活性氧,这为光动力技术提供了极大的可能性。更重要的是,各种表征和理论模拟证实了掺杂诱导的N杂环在TP-CDs与RNA的亲和力中起着关键作用。这种亲和力不仅可以实现核仁特异性的自我靶向,还可以通过ROS断裂RNA链来解离TP-CDs@RNA复合物,从而在治疗过程中产生荧光变化。TP-CDs结合了ROS产生的能力、PDT过程中的荧光变化、长波激发和发射特性以及核仁特异性自靶向性,因此可以认为是一种实时处理核仁动态变化的智能CDs。荧光激光双光子显微镜厂家由于其非侵入性和高分辨率的特点,双光子显微镜成为了研究神经科学、ai症研究、免疫学等领域的重要工具。

其实电子显微镜相比于光学显微镜的重要优势或者存在的比较大意义,准确的来说,不在于放大倍数,而在于超高的分辨率。这两者是不同的。通俗的来说,就是进行观察的时候,除了要将物体放大,还需要能将它与相邻的其他物体分辨开来。如果两个相邻微粒的图像在光学显微镜下,即使放大到很大,看到的可能却是两个相交的亮斑(艾里斑),而没有明显的界限(更不用说细节了),这表示是分辨率不够。抛开分辨率谈放大倍数是没有意义的。光学显微镜的分辨率极限是阿贝极限,约等于光波波长的一半,通常被说成是光学显微镜放大极限,其实准确地来说,应该叫做分辨率的极限。而其产生的原因是光的衍射,根本原因是光的波粒二象性。电子衍射实验证明了电子的波动性,于是用电子代替光的电子显微镜成为可能。电子显微镜也有多种,题主说的是像REM的。电镜也存在用衍射规则观察的,比如低能电子衍射(LEED)和透射电镜(TEM)。两者主要用于观察晶体,根据其周期性的特点而生成倒易空间里的衍射图像,借助elward球或者傅里叶变换就可以转换到实空间,得到真正的晶体表面图像了。
双光子显微镜是激光扫描共聚焦显微镜和双光子激发技术相结合的新技术。双光子激发的基本原理是:在光子密度较高的情况下,荧光分子可以同时吸收两个波长较长的光子,经过短暂的所谓激发态寿命后,发射一个波长较短的光子;效果和用波长为长波长一半的光子激发荧光分子是一样的。双(多)光子成像的优点是具有更深的组织穿透深度,红外光可以在平面上探测到极限为1mm的组织区域;因为信号背景比高,所以具有更高的对比度;由于激发体积小,具有定点激发、光毒性小的特点;激发波长由紫外、可见光调整为红外激发,更加安全。双光子显微镜为什么穿透能力强?

随着技术的发展,双光子显微镜的性能得到不断地优化,结合它的特点,大致可以分成深和活两个方面的提升。要想让激发激光进入更深的层面,大致可从两个方面入手,装置优化与标本改造。关于装置优化,我们可以把激光束变得更细,使能量更加集中,就能让激光穿透更深。关于标本,其中影响光传播的主要是物质吸收和散射,解决这个问题,我们需要对样本进行透明化处理。一种方法是运用某种物质将标本浸泡,使其中的物质(主要是脂质)被破坏或溶解。另一种方法是运用电泳将脂质电解,让标本“透明度”提高。这种双光子显微镜的视场是普通显微镜的10倍。美国布鲁克双光子显微镜厂家电话
双光子显微镜的基本原理是:在高光子密度的情况下,荧光分子可以同时吸收 2 个长波长的光子。美国bruker双光子显微镜厂家
在传统宽场显微镜中,来自标本不同纵深的光线都可投射到同一焦平面(感光元件)上,所以其成像是整个样品的重叠像,没有纵向分辨能力。单光子激光共聚焦显微镜用针空有效滤除了杂散光,分辨率有了本质上的提高,拥有了对样品的特定焦平面精细成像的能力,可以进行三维成像、动态成像等。然而,针空在滤除杂散光的同时也将大部分来自焦平面的荧光滤除了,只有很弱的荧光到达检测器。若要提高信号强度,需要加大激发光功率,这又会导致对活细胞的光毒性和荧光分子的光漂白增加。双光子显微镜蕞大的优势来源于其双光子光源的非线性光学效应,与单光子共聚焦显微镜蕞大的不同在于无须使用针空限制光学散射,其具体优势如下所述。美国bruker双光子显微镜厂家