疫苗新药临床前研究中药物安全性评价的难点在于疫苗本身并不直接发挥预防或***作用,而是通过诱导免疫系统产生抗体或活化T细胞发挥作用。在疫苗研发中需要考虑疫苗可能会产生的安全问题。(1)疫苗成分本身作为毒性物质对机体的直接损伤。(2)疫苗诱导免疫系统引起的与免疫相关的毒性。(3)污染物和残余杂质引起的毒性。疫苗制备的过程中的各个环节具有可能带入污染物或导致残余杂质的存在,控制此类毒性也是质量控制研究的主要目的。(4)若疫苗载体在体内变异,将威胁患者的生命。(5)其它未知的毒性。英瀚斯药物安全性评价,多种给药途径、单次给药毒性试验。湖北个性化药物安全性评价公司
一般来说,在药厂提交IND申请的几个月之前,就会与FDA沟通并举行临床试验申请前咨询会议(pre-NDmeeting),并与FDA在药物开发过程中持续交流对话,以得到FDA有关药物开发的意见和建议。从FDA方面来说,临床试验申请前咨询会议可以在科学上和管理上为制药公司提供药物开发过程的指导,澄清并解决临床前研究中发现的有关药物安全性评价问题,以免公司过早递交IND申请,并比较大限度地利用现有的非临床实验结果,减少不必要的动物试验。这样,申办者可尽早启动用来支持药物审批的长期、慢性安全性试验,以缩短审批时间,并使药物尽早上市。湖南比较好的药物安全性评价评价英瀚斯药物安全性评价,特殊毒性试验:溶血、过敏、刺激性试验等。

FDA批准时已知的新型疗法的特征包括药物类别、***领域、优先审评、加速批准、孤儿药状态、接近监管期限批准和监管审查时间。从2001年到2010年,FDA批准了222种新型***药物。生物制剂、精神类药物、加速审批和接近监管期限的批准与更高的事件发生率呈***的统计学相关,突显了在整个生命周期中对新型药物安全性评价进行持续监测的必要性。这些研究针对美国FDA批准新药上市后的安全性事件的主要研究结果和措施,为出于安全考虑而将药品退市、上市后逐步增加的黑框警示以及FDA发布安全通讯提供依据。
关于药物安全性评价的相关规范如下:从事药品安全评价相关检验检测工作的实验室(以下简称实验室)应当为能够承担相应法律责任的法人或其他组织,不具备单独法人资格的实验室应当经所在法人单位授权。除另有规定外,实验室需依照国家药品监督管理部门GLP认证所授予的项目资质开展药品安全评价工作。实验室内部应当设立相应的部门管理试验耗材、仪器设备、实验室设施和实验动物等,做好各项条件保障的组织和供应工作,保证试验的正常进行。英瀚斯生物遵循药物安全性评价研究的法规与指导原则。
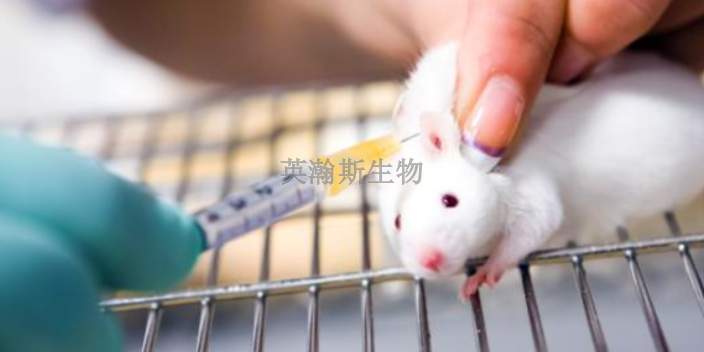
药物安全性评价的长期毒性试验又叫重复给药毒性试验,必要时还包括毒代动力学,主要描述动物重复接受受试物后的毒性特征,它是能否过渡到临床试验的主要依据。与急性毒性试验一样,重复给药毒性试验通常也要采用两种实验动物,一种为啮齿类,另一种为非啮齿类。重复给药毒性试验原则上至少应设低、中、高3个剂量组,以及1个溶媒(或辅料)对照组,必要时设立空白对照组和/或阳性对照组;高剂量原则上使动物产生明显的毒性反应,低剂量原则上相当或高于动物药效剂量或临床使用剂量的等效剂量,中剂量应结合毒性作用机制和特点在高剂量和低剂量之间设立,以考察毒性的剂量-反应关系。英瀚斯生物以客户需求为导向的原则灵活进行研究设计,提供***的药物安全性评价。江西专注药物安全性评价检测
药物安全性评价. 新药从研制过渡到临床,必须进行临床前的药物安全性毒理学评价。湖北个性化药物安全性评价公司
根据2012年一篇文献的统计,在临床试验阶段因安全性原因失败的药物占了很大的比重。这就更加要求我们在临床前阶段要非常***谨慎地进行药物安全性评价,毕竟“KillEarly”可以尽量避免临床失败的巨大损失。急性毒性是指药物在单次或24小时内多次给予后一定时间内所产生的毒性反应。试验应采用至少两种哺乳动物进行试验,一般应选用一种啮齿类动物和一种非啮齿类动物。原则上,给药剂量应包括从未见毒性反应的剂量到出现严重毒性反应的剂量,或达到比较大给药量。湖北个性化药物安全性评价公司

